

Since 1994, medical devices have had to comply with the requirements of European Directives 93/42/EEC and 90/385/EEC. Conformity with this regulation allows the affixing of the CE marking, enabling the free circulation of medical devices within the European Union (EU).
The European Union has recently revised this regulation through the European Regulation 2017/745/EU for medical devices (MDR), which finally came into effect on May 26, 2021. The adoption of a regulation instead of a directive ensures uniformity in the regulatory framework through its direct application.
This new regulation imposes new responsibilities on economic operators involved in the supply chain of medical devices: manufacturer, authorized representative, importer, distributor.
What are the main changes introduced by this new regulation (MDR)?
– Redesignation of Notified Bodies (NB) under MDR and EU control of Notified Bodies (NB)
– Improved traceability through the Unique Device Identification (UDI)
– Inclusion of certain aesthetic devices with the same characteristics and risk profile as medical devices covered by the regulation
– Enhanced transparency through EUDAMED, the medical device database
– Strengthened requirements for clinical data and post-market surveillance
What transition periods will apply?
Initially, the MDR was to come into effect on May 26, 2020. Due to the COVID-19 health crisis in 2020, the EU decided to postpone this date through Regulation (EU) 2020/561.
The implementation date was thus postponed to May 26, 2021, with a transition period for manufacturers with medical devices already on the European market (“LEGACY DEVICES”) until May 26, 2024 (end of the issuance of certificates under the directive) and the availability of these medical devices on the market until May 26, 2025. However, for class I medical devices and new medical devices entering the European market, they had to comply with the MDR since May 26, 2021.
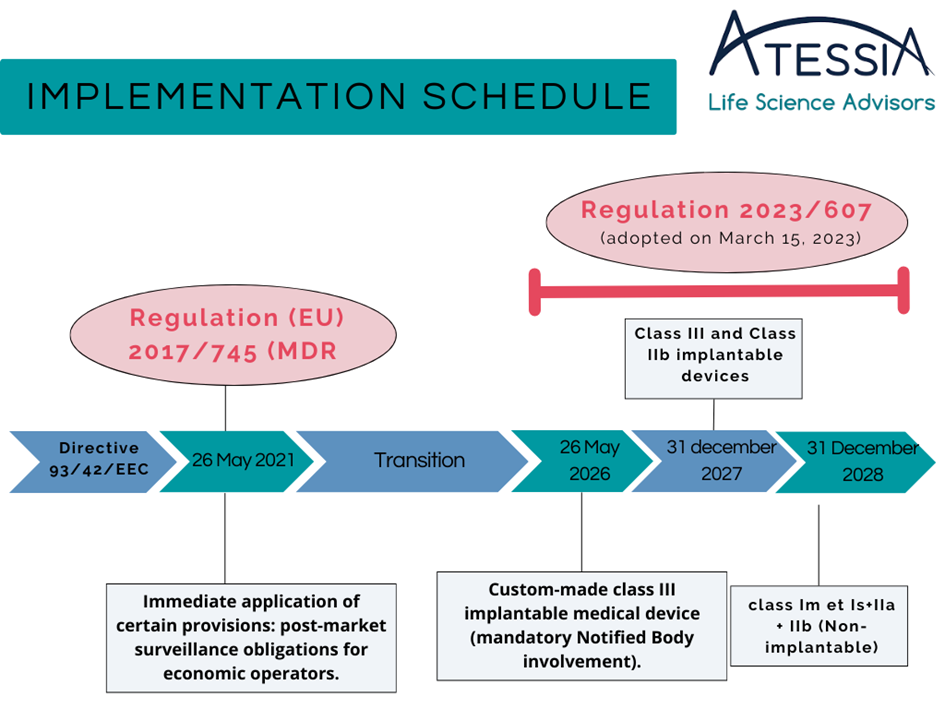
On March 20, 2023, the EU assessed a potential shortage risk for many medical devices and adopted Regulation EU 2023/607, amending Regulation (EU) 2017/745 concerning transitional provisions (Article 120).
The extension dates of certificates under Directive 93/43/EEC are extended based on the class of the medical device:
However, manufacturers covered by this extension must meet the following conditions:
1/ The devices continue to comply with Directive 90/385/EEC or Directive 93/42/EEC, as applicable. ;
2/ There are no significant changes to the design and intended use.;
3/ The devices do not present an unacceptable risk to the health or safety of patients, users, or other persons, considering other aspects related to public health protection. ;
4/ no later than 26 May 2024, the manufacturer has put in place a quality management system in accordance with Article 10(9); ;
5/ The manufacturer or authorized representative has lodged a formal conformity assessment request under the MDR for a medical device or a replacement device to a Notified Body by September 26, 2024, and the Notified Body and the manufacturer have signed a written agreement.
Special case for custom-made class III implantable devices – transitional period introduced:
The conformity assessment of custom-made class III implantable devices requires an assessment by a Notified Body.
Custom-made class III implantable medical devices can be placed on the market without the corresponding certificate until May 26, 2026, provided that the manufacturer has submitted an application to a designated Notified Body under the MDR before May 26, 2024, and has signed a contract with that Notified Body before September 26, 2024.
MDR requirements applicable from May 26, 2021:
However, it is important to note that certain MDR requirements shall apply to devices since May 26, 2021, such as requirements related to post-market surveillance, market surveillance, vigilance, and the registration of economic operators.
Removal of the availability date:
Additionally, Regulation EU 2023/607 completely removes the availability deadline, allowing “LEGACY DEVICES” to be available without a deadline (still respecting the device’s usage limit date). This removal also applies to in vitro diagnostic medical devices marketed before May 26, 2022, in Regulation (EU) 2017/746 (IVDR).
Are CE certificates under the directives still valid during this extension period?
Certificates that expired before the entry into force of Regulation 2023/607 (March 20, 2023) should only be considered valid if:
– Either before the certificate’s expiration date, the manufacturer and Notified Body signed a conformity assessment agreement before the certificate’s expiration date.
– Or if a competent authority has granted a derogation.
Notified Bodies can no longer issue or modify CE certificates under the directives since May 26, 2021. Therefore, they are considered extended unless they have been withdrawn.
Manufacturers can issue a self-declaration confirming that they comply with the extension conditions, indicating the end date of the transition period (the medical devices covered by the extension must be clearly specified). At the manufacturer’s request, the Notified Body can also issue a confirmation letter.
It is important to remember that despite these delays, manufacturers must now act toward this new regulation. The submission period to Notified Bodies can take from 6 to 18 months, depending on the Notified Body and the medical device.
Atessia supports its clients through all stages of MDR compliance.