What future for therapeutic cannabis in France?
A bit of history
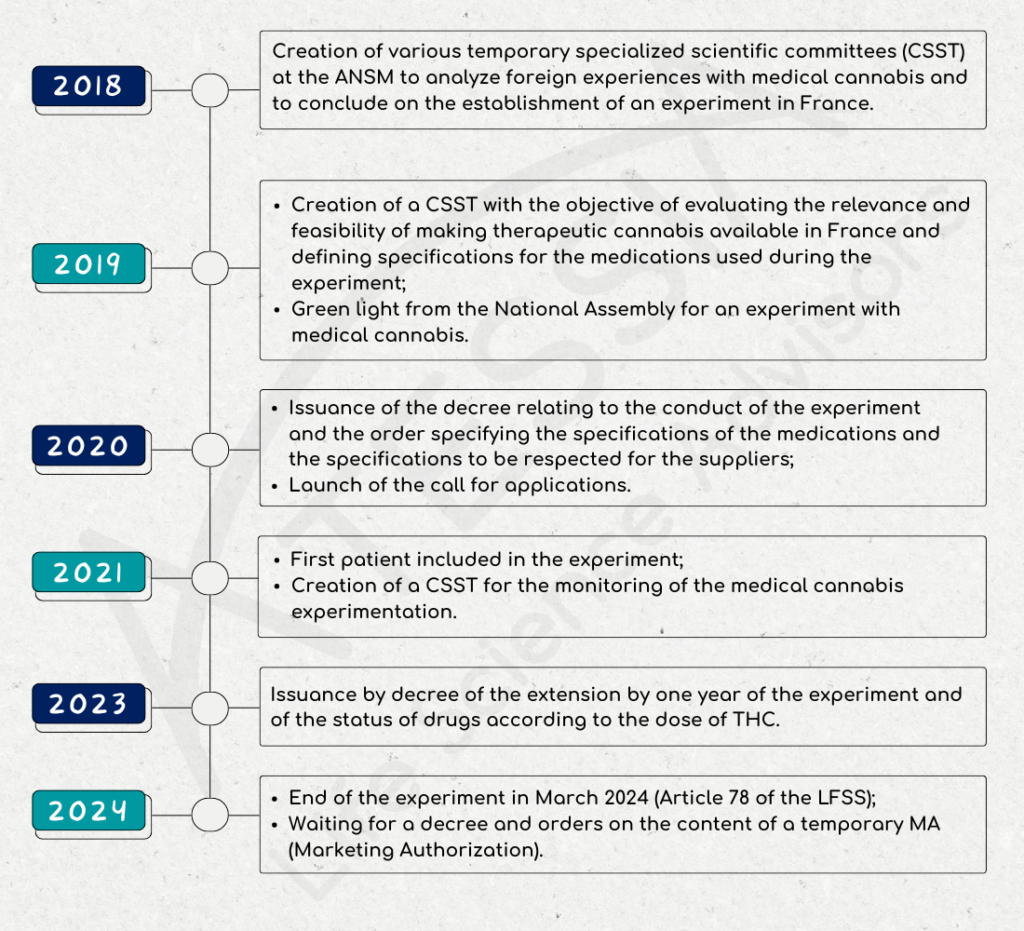
In September 2018, a multidisciplinary scientific committee made up of ANSM, healthcare professionals and patients was set up to review scientific knowledge and foreign experience on medical cannabis.
In December 2018, this committee concludes that the use of cannabis is appropriate for patients in certain clinical situations, and wishes to set up an experiment.
As of January 2019, a temporary specialized scientific committee (CSST) is being set up with the aim of assessing the relevance and feasibility of making therapeutic cannabis available in France. This CSST is tasked with issuing an opinion on:
- the therapeutic value of cannabis in the treatment of certain pathologies;
- the modalities for making cannabis available for medical use.
At the same time, the committee had to define specifications for:
- The drugs used during the experiment;
- The content of training for doctors and pharmacists and information for patients;
- The content of the patient follow-up register.
Launch of the experimental framework
On October 25, 2019, the French National Assembly gives the go-ahead for an experiment in the use of medical cannabis.
Decree no. 2020-1230 of October 7, 2020 on the experimentation of the medical use of cannabis defined in particular:
- the duration of the pilot (2 years),
- the status of medical cannabis as a narcotic drug,
- the number of patients who may be included,
- the conditions of treatment,
- the setting up of a register to monitor adverse events.
The Order of October 16, 2020 set out the specifications for cannabis-based medicines used during the experiment provided for in Article 43 of Law no. 2019-1446 of December 24, 2019 on the financing of social security (LFSS) for 2020, together with the conditions for making them available and the therapeutic indications or clinical situations in which they will be used.
This order defined:
- The indications for which medical cannabis products entered the trial,
- The authorized pharmaceutical forms:
- form for inhalation by vaporization, such as dried flowering tops or granules;
- oral form in capsule or equivalent form
- oral or sublingual oil form
- as well as specifications for the free supply and distribution of cannabis-based medicines for patients taking part in the experiment in the medical use of cannabis.
The call for applications launched on October 19, 2020 by ANSM closed on November 24, 2020. Applications were examined on the basis of strict and demanding specifications in terms of compliance with good cultivation and manufacturing practices, drug quality and securing the distribution circuit as defined in the decree. This examination was carried out by the ANSM, and in particular by its control laboratories, and by experts from the Temporary Specialized Scientific Committee (CSST).
In all, six supplier/operator pairs were selected for the trial.
First patient – Start of the trial
On March 26, 2021, the medical cannabis trial officially began with the inclusion of the first patient at Clermont-Ferrand University Hospital. The experiment will run for 2 years.
In June 2021, a CSST was set up to monitor the medical cannabis experiment. It is made up of 16 members, including 4 patients and healthcare professionals, general practitioners, specialists in the therapeutic indications selected for medical cannabis, pharmacists and representatives of the Centre Régional de Pharmacovigilance (CRPV) and the Centres d’Evaluation et d’Information sur la Pharmacodépendance-addictovigilance (CEIP-A).
This committee is involved in monitoring the progress of the experiment and must issue an opinion on the evaluation data collected and on the framework for the marketing and use of medical cannabis.
2023: the first turning point
Decree 2023-202 of March 25, 2023, amending Decree 2020-1230 of October 7, 2020:
- extends the trial period for the medical use of cannabis by one year;
- indicates that medicines with a THC content of over 0.30% are subject to the narcotics regime; conversely, those with a THC content of less than or equal to 0.30% are now subject to the regime for medicines covered by lists I and II of poisonous substances.
At the same time, various decrees dated March 25, 2023 specify that:
- Pharmacovigilance and addictovigilance will now be handled in the same way as for other drugs;
- Inhalation granules have been discontinued;
- The ANSM is no longer responsible for selecting suppliers and operators of cannabis for medical use. The Direction Générale de la Santé (DGS) is now the competent authority in this area, via a public procurement contract. The medicines used will therefore no longer be supplied free of charge by participating companies.
End of the experiment
Article 78 of the Social Security Finance Act of December 26, 2023 puts an end to the experiment.
As of March 26, 2024, no new patients can be included. Patients included before this date will still be able to benefit from their treatment, with the exception of inhaled forms.
Other patients will have to wait until a medicine is authorized and available “no later than December 31, 2024” before they can benefit from a cannabis-based treatment for therapeutic use.
What’s next?
Article 78 creates an ad hoc status for cannabis for medical use: pending the granting of marketing authorizations (MA) in due form, the use of cannabis-based medicines may be authorized by the ANSM in the form of temporary MA, for a temporary period of 5 years, renewable. Such authorization may only be granted if the use of these products meets the special needs of a given patient, and there is no suitable pharmaceutical packsize available, including due to the absence of effective marketing, with, for example, a marketing authorization (article L5121-1 4° of the CSP).
Article written by Isabelle BARBIEUX, Senior Quality Assurance Advisor
Are you wondering? Want to get started?
Atessia is here to answer your questions and help you make your project a reality.
Call on our experts.