

Quel avenir pour le cannabis thérapeutique en France ?
Un peu d’histoire
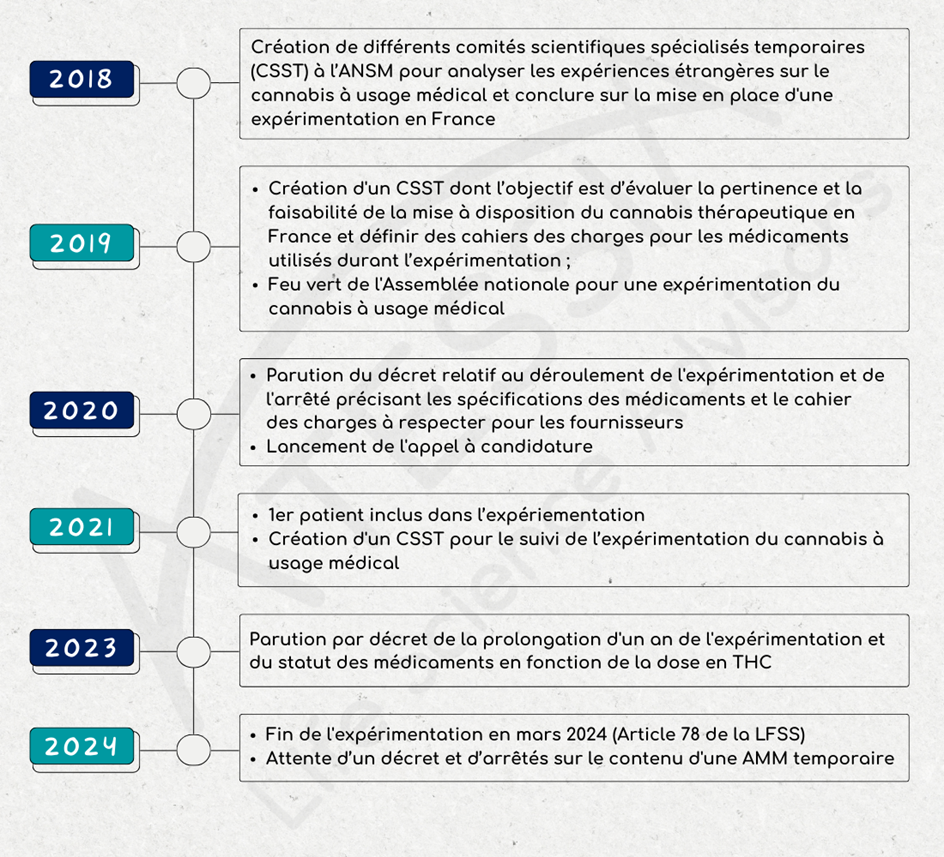
En septembre 2018, un comité scientifique pluridisciplinaire formé de l’ANSM, de professionnels de santé et de patients, est créé pour examiner les connaissances scientifiques et les expériences étrangères sur le cannabis à usage médical.
En décembre 2018, ce comité conclut à la pertinence de l’usage du cannabis pour les patients dans certaines situations cliniques et souhaite mettre en place une expérimentation.
A partir de janvier 2019, un comité scientifique spécialisé temporaire (CSST) se crée dont l’objectif est d’évaluer la pertinence et la faisabilité de la mise à disposition du cannabis thérapeutique en France. Ce CSST est chargé de rendre un avis sur :
- l’intérêt thérapeutique du cannabis pour le traitement de certaines pathologies ;
- les modalités de mise à disposition du cannabis dans le cadre d’une utilisation médicale.
Ce comité devait définir, en parallèle, des cahiers des charges pour :
- Les médicaments utilisés durant l’expérimentation ;
- Le contenu de la formation destinée aux médecins et pharmaciens et le contenu de l’information aux patients ;
- Le contenu du registre de suivi des patients.
Lancement du cadre de l’expérimentation
Le 25 octobre 2019 l’Assemblée nationale donne son feu vert à une expérimentation de l’usage du cannabis médical.
Le décret n° 2020-1230 du 7 octobre 2020 relatif à l’expérimentation de l’usage médical du cannabis a défini notamment :
- la durée de l’expérimentation (2 ans),
- le statut du cannabis à usage thérapeutique comme médicament stupéfiant
- le nombre de patients pouvant être inclus,
- les conditions de prise en charge,
- la mise en place d’un registre pour le suivi des évènements indésirables.
L’arrêté du 16 octobre 2020 a fixé les spécifications des médicaments à base de cannabis utilisés pendant l’expérimentation prévue à l’article 43 de la Loi n°2019-1446 du 24 décembre 2019 de financement de la sécurité sociale (LFSS) pour 2020, les conditions de leur mise à disposition ainsi que les indications thérapeutiques ou situations cliniques dans lesquelles ils seront utilisés.
Cet arrêté définissait :
- Les indications pour lesquelles les produits à base de cannabis médical entraient dans l’expérimentation,
- Les formes pharmaceutiques autorisées :
- forme pour inhalation par vaporisation telle que des sommités fleuries séchées ou des granulés ;
- forme orale sous forme de capsule ou forme équivalente
- forme orale ou sublinguale sous forme d’huile
- ainsi qu’un cahier des charges pour la fourniture et la distribution à titre gratuit de médicaments à base de cannabis pour les patients qui participeront à l’expérimentation de l’usage médical du cannabis.
L’appel à candidature lancé le 19 octobre 2020 par l’ANSM a été clôturé le 24 novembre 2020. L’examen des candidatures a été réalisé sur la base d’un cahier des charges strict et exigeant en termes de respect des bonnes pratiques de culture et de fabrication, de qualité des médicaments et de sécurisation du circuit de distribution comme défini dans l’arrêté. Cet examen a été réalisé par l’ANSM et en particulier par ses laboratoires de contrôle, et des experts du comité scientifique spécialisé temporaire (CSST).
Au total, six binômes fournisseurs/exploitants ont été retenus pour l’expérimentation.
Premier patient – Démarrage de l’expérimentation
Le 26 mars 2021, l’expérimentation du cannabis à usage médical a officiellement débuté avec l’inclusion du premier patient au CHU de Clermont-Ferrand. L’expérimentation est lancée pour 2 ans.
En juin 2021, un CSST est créé pour le suivi de l’expérimentation du cannabis à usage médical. Il est composé de 16 membres, dont 4 patients et des professionnels de santé, médecins généralistes, spécialistes des indications thérapeutiques retenues pour le cannabis médical, pharmaciens et représentants des Centre régionaux de pharmacovigilance (CRPV) et des Centres d’évaluation et d’information sur la pharmacodépendance-addictovigilance (CEIP-A).
Ce comité participe à la surveillance du déroulement de l’expérimentation et doit rendre un avis sur les données d’évaluation recueillies et sur l’encadrement de la mise sur le marché et de l’utilisation du cannabis médical.
2023 : premier virage
Le décret 2023-202 du 25 mars 2023 modifiant le décret 2020-1230 du 7 octobre 2020 :
- prolonge la durée de l’expérimentation de l’usage médical du cannabis d’un an ;
- indique que les médicaments avec une teneur en THC supérieure à 0,30% sont soumis au régime des médicaments stupéfiants ; à l’inverse, ceux avec une teneur en THC inférieure ou égal à 0,30% sont désormais soumis au régime des médicaments relevant des listes I et II des substances vénéneuses.
En parallèle, différents arrêtés du 25 mars 2023 précisent notamment que :
- La pharmacovigilance et l’addictovigilance seront désormais assurées de la même façon que pour les autres médicaments ;
- Les granulés pour inhalation sont supprimés ;
- L’ANSM n’est plus responsable de la sélection des fournisseurs et des exploitants de cannabis à usage médical. C’est la Direction générale de la santé (DGS) qui est désormais l’autorité compétente en la matière via un marché public. Les médicaments utilisés ne seront donc plus fournis à titre gratuit par les entreprises participantes.
Fin de l’expérimentation
L’article 78 de la loi de finance de la sécurité sociale du 26 décembre 2023 met un terme à l’expérimentation.
À compter du 26 mars 2024, aucune inclusion de patient n’est possible. Les patients inclus avant cette date pourront toujours bénéficier de leur traitement à l’exception des formes inhalées.
Les autres patients devront attendre qu’un médicament soit autorisé et disponible « au plus tard jusqu’au 31 décembre 2024 » pour pouvoir bénéficier d’un traitement à base de cannabis à usage thérapeutique.
Et après :
L’article 78 crée un statut ad hoc pour le cannabis à usage médical : dans l’attente de l’octroi d’autorisations de mise sur le marché (AMM) en bon et due forme, l’utilisation de médicaments à base de cannabis pourra être autorisée par l’ANSM sous la forme d’AMM temporaires, pour une période temporaire de 5 ans, renouvelable. Une telle autorisation ne pourra être délivrée que si l’utilisation de ces produits répond aux besoins spéciaux d’un patient déterminé et qu’il n’existe pas de spécialité pharmaceutique disponible et adaptée, y compris du fait de l’absence de commercialisation effective, disposant par exemple, d’une autorisation de mise sur le marché (article L5121-1 4° du CSP).
Article rédigé par Isabelle BARBIEUX, Consultant Sénior Assurance Qualité
Vous vous interrogez ? Vous souhaitez vous lancer ?
Atessia est là pour vous répondre à vos interrogations et vous aider à concrétiser votre projet.
Faites appel à nos experts.









{kind=link}