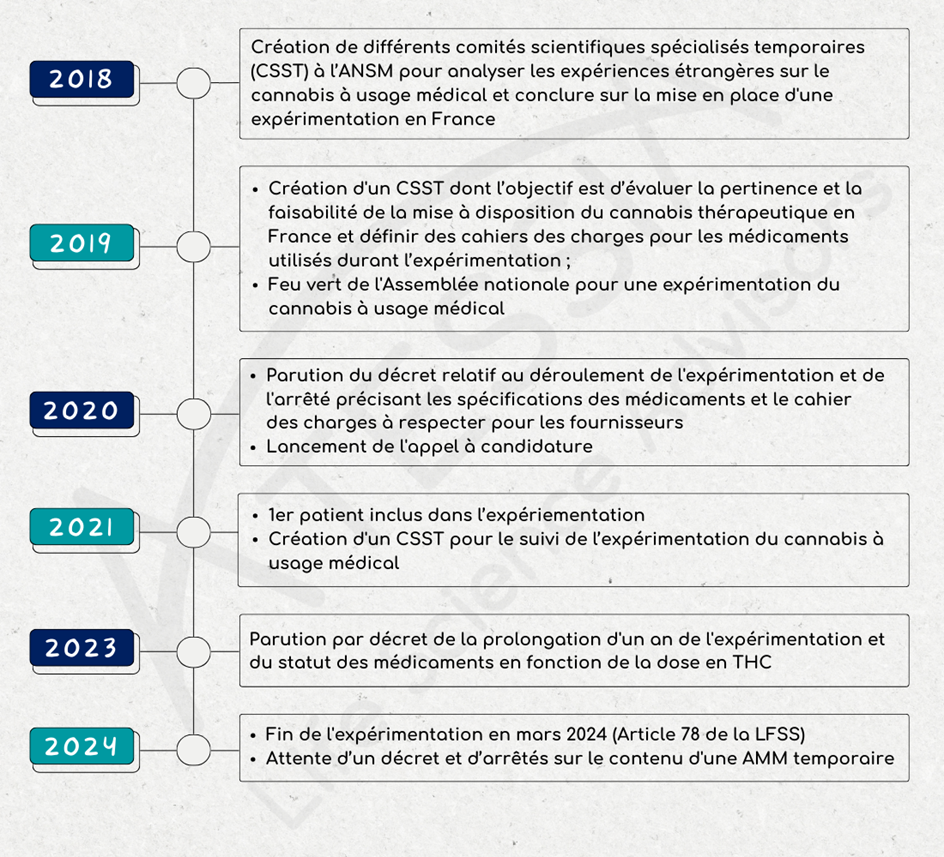
Protocole de gestion des changements post-approbation «PACMP»
En Europe, les changements apportés à une autorisation de mise sur le marché (AMM) de médicament humain sont couverts par le règlement (CE) N°1234/2008 du 24 novembre 2008. Ce règlement est applicable depuis le 1er janvier 2010 aux AMM obtenues en procédures centralisées, décentralisées et de reconnaissance mutuelle, et depuis le 4 août 2013 aux AMM obtenues en procédures nationales. Il présente de multiples possibilités permettant de classer les modifications pouvant être apportées à une AMM dont le protocole de gestion des modifications après autorisation (connu sous l’acronyme anglais « PACMP » pour Post-Approval Change Management Protocol ») auquel nous allons nous attacher dans cet article.
- Origine et définition
Un PACMP est un outil réglementaire offrant prévisibilité et transparence en termes d’exigences et d’études nécessaires à la mise en œuvre d’un changement strictement CMC (pour « Chemistry, Manufacturing & Controls »), car le protocole des changements envisagés approuvé constitue un accord entre le titulaire de l’AMM et l’autorité réglementaire. Il s’agit d’une approche par étapes de l’évaluation des modifications permettant dans un 1er temps, une évaluation précoce de la stratégie de modification et, dans un 2nd temps, une évaluation distincte ultérieure des données produites après mise en œuvre des changements envisagés sur la base de la stratégie convenue.
L’objectif visé par cet outil est de permettre une mise en œuvre plus rapide et plus prévisible des modifications après approbation, étant donné que le titulaire de l’AMM aura obtenu au préalable l’accord des autorités sur la stratégie proposée et les tests permettant de vérifier l’effet de la modification sur la qualité du produit. En règle générale, la catégorie de variation désignée pour signaler les modifications dans le cadre d’un PACMP est inférieure d’au moins une catégorie à ce qu’elle serait normalement (ex. type IB au lieu de type II). La mise en œuvre des changements prévus dans un PACMP approuvé est donc plus rapide et moins risquée pour les laboratoires qui en font la demande, ce qui bénéficie in fine à la commercialisation du médicament et donc au patient.
En 2012, l’EMA a regroupé un ensemble de questions-réponses concernant le PACMP dans le document « Questions and answers on post approval change management protocols (EMA/CHMP/CVMP/QWP/586330/2010) ». Les éléments suivants y sont abordés :
- Suggestions de contenu du PACMP ;
- Mécanismes de soumission et d’évaluation du PACMP ;
- Mise en œuvre du/des changements après finalisation des études décrites dans le PACMP ;
- Types de changements pouvant faire l’objet d’un PACMP ;
- Changements multiples dans le cadre d’un PACMP unique ;
- Place du PACMP dans le Module 3 de l’AMM ;
- PACMP et développement selon l’approche traditionnelle versus l’approche améliorée (dite « QbD » pour « Quality by Design »).
En 2019, la ligne directrice ICH Q12 « Technical and regulatory considerations for pharmaceutical product lifecycle management », proposée en vue de fournir un cadre visant à faciliter la gestion des changements apportés aux propriétés chimiques, à la fabrication et aux mesures de contrôles après l’autorisation, de manière plus prévisible et plus efficace tout au long du cycle de vie du produit, est venue renforcer l’encrage du PACMP comme outil réglementaire incontournable dans la gestion du cycle de vie des médicaments en Europe.
- Périmètre de l’utilisation du PACMP
Le PACMP concerne aussi bien des modifications CMC relatives à la substance active (SA) que des modifications relatives au produit fini (PF). Il s’applique à tous les médicaments à usage humain et vétérinaire, y compris les produits biotechnologiques ou biologiques, qu’une approche traditionnelle ou améliorée (« QbD ») ait été suivie pour le développement du produit. Son utilisation est toutefois facultative.
Un PACMP peut être appliqué à un seul produit, à plusieurs produits ou à plusieurs produits et à plusieurs sites.
La présence d’un PACMP dans son dossier d’AMM implique une analyse de risque minutieuse et une compréhension totale des différentes évaluations de risque afin de garantir que la qualité, la sécurité et l’efficacité du médicament ne sont jamais compromises.
Il est à noter qu’aucune modification décrite dans un PACMP ne doit entraîner de risques supplémentaires pour la sécurité des patients, la qualité ou l’efficacité du produit. Aussi, une modification CMC qui nécessiterait des données d’efficacité, de sécurité ou de pharmacocinétique/de pharmacodynamique humaines pour évaluer l’effet de la modification (ex. certaines modifications de la formulation, des études cliniques ou non cliniques pour évaluer de nouvelles impuretés, l’évaluation de l’immunogénicité ou de l’antigénicité) ne peut pas être incluse dans un PACMP.
- Matérialisation du PACMP dans le dossier d’AMM
Le PACMP se matérialise sous la forme d’un ou plusieurs document(s) présenté(s) en section 3.2.R « Regional Information » du dossier d’AMM. Il peut être prévu dès la demande d’AMM ou bien au cours d’une demande de variation (Type II) de l’AMM.
- Modifications d’AMM en lien avec le PACMP
Les différentes variations en lien avec l’introduction, la suppression ou encore la modification d’un PACMP dans un dossier d’AMM sont présentés dans le tableau ci-contre.
| Substance active | Produit fini |
| Variation Type II/B.I.e.2 : Introduction d’un protocole de gestion des modifications après autorisation relatif à la substance active > Absence de conditions à remplir. > Trois éléments supportifs à verser : 1. Description détaillée de la modification proposée. 2. Protocole de gestion des modifications relatif à la substance active. 3. Version modifiée de la/des sections concernées du dossier. | Variation Type II/B.II.g.2 : Introduction d’un protocole de gestion des modifications après autorisation relatif au produit fini > Absence de conditions à remplir. > Trois éléments supportifs à verser : 1. Description détaillée de la modification proposée. 2. Protocole de gestion des modifications relatif au produit fini. 3. Version modifiée de la/des sections concernées du dossier. |
| Variation Type IAIN/B.I.e.3 : Suppression d’un protocole approuvé de gestion des modifications relatif à la substance active > Une condition à remplir : 1. La suppression du protocole approuvé de gestion des modifications relatif à la substance active n’est pas la conséquence d’événements imprévus ni de résultats en dehors des spécifications au cours de la mise en œuvre de la ou des modifications décrites dans le protocole et n’a pas d’incidence sur les informations déjà approuvées contenues dans le dossier. > Deux éléments supportifs à verser : 1. Justification de la suppression proposée. 2. Version modifiée de la/des sections concernées du dossier. | Variation Type IAIN/B.II.g.3 : Suppression d’un protocole approuvé de gestion des modifications relatif au produit fini > Une condition à remplir : 1. La suppression du protocole approuvé de gestion des modifications relatif au produit fini n’est pas la conséquence d’événements imprévus ni de résultats en dehors des spécifications au cours de la mise en œuvre de la ou des modifications décrites dans le protocole et n’a pas d’incidence sur les informations déjà approuvées du dossier. > Deux éléments supportifs à verser : 1. Justification de la suppression proposée. 2. Version modifiée de la/des sections concernées du dossier. |
| Variation Type II/B.I.e.4a) : Changements apportés à un protocole approuvé de gestion des modifications – Changements majeurs apportés à un protocole approuvé de gestion des modifications | Variation Type II/B.II.g.4a) : Changements apportés à un protocole approuvé de gestion des modifications – Changements majeurs apportés à un protocole approuvé de gestion des modifications |
| Variation Type IB/B.I.e.4b) : Changements apportés à un protocole approuvé de gestion des modifications – Changements mineurs apportés à un protocole approuvé de gestion des modifications, qui n’ont pas d’incidence sur la stratégie définie dans le protocole > Absence de conditions à remplir. > Un élément supportif à verser : 1. Déclaration selon laquelle tout changement reste dans le cadre des limites actuellement approuvées. Autre déclaration selon laquelle l’évaluation de la comparabilité n’est pas requise pour les médicaments biologiques/immunologiques. | Variation Type IB/B.II.g.4b) : Changements apportés à un protocole approuvé de gestion des modifications – Changements mineurs apportés à un protocole approuvé de gestion des modifications, qui n’ont pas d’incidence sur la stratégie définie dans le protocole > Absence de conditions à remplir. > Un élément supportif à verser : 1. Déclaration selon laquelle tout changement reste dans le cadre des limites actuellement approuvées. Autre déclaration selon laquelle l’évaluation de la comparabilité n’est pas requise pour les médicaments biologiques/immunologiques. |
| Variation Type IAIN/B.I.e.5a) : Mise en œuvre de changements prévus dans un protocole approuvé de gestion des modifications – La mise en œuvre du changement ne nécessite pas d’informations complémentaires > Une condition à remplir : 1. Le changement proposé a été effectué en parfaite conformité avec le protocole approuvé de gestion des modifications. > Trois éléments supportifs à verser : 1. Référence au protocole approuvé de gestion des modifications. 2. Déclaration contenant les informations suivantes : le changement est en conformité avec le protocole approuvé de gestion des modifications et les résultats de l’étude répondent aux critères d’acceptation indiqués dans le protocole. Autre déclaration selon laquelle l’évaluation de la comparabilité n’est pas requise pour les médicaments biologiques/immunologiques. 4. Version modifiée de la/des sections concernées du dossier. | Variation Type IAIN/B.II.g.5a) : Mise en œuvre de changements prévus dans un protocole approuvé de gestion des modifications – La mise en œuvre du changement ne nécessite pas d’informations complémentaires > Une condition à remplir : 1. Le changement proposé a été effectué en parfaite conformité avec le protocole approuvé de gestion des modifications et doit donc être notifié immédiatement après sa mise en œuvre. > Trois éléments supportifs à verser : 1. Référence au protocole approuvé de gestion des modifications. 2. Déclaration contenant les informations suivantes : le changement est en conformité avec le protocole approuvé de gestion des modifications et les résultats de l’étude répondent aux critères d’acceptation indiqués dans le protocole. Autre déclaration selon laquelle l’évaluation de la comparabilité n’est pas requise pour les médicaments biologiques/immunologiques. 4. Version modifiée de la/des sections concernées du dossier. |
| Variation Type IB/B.I.e.5b) : Mise en œuvre de changements prévus dans un protocole approuvé de gestion des modifications – La mise en œuvre du changement nécessite des informations complémentaires > Absence de conditions à remplir. > Quatre éléments supportifs à verser : 1. Référence au protocole approuvé de gestion des modifications. 2. Déclaration contenant les informations suivantes: le changement est en conformité avec le protocole approuvé de gestion des modifications et les résultats de l’étude répondent aux critères d’acceptation indiqués dans le protocole. Autre déclaration selon laquelle l’évaluation de la comparabilité n’est pas requise pour les médicaments biologiques/immunologiques. 3. Résultats des études menées en conformité avec le protocole approuvé de gestion des modifications. 4. Version modifiée de la/des sections concernées du dossier. | Variation Type IB/B.II.g.5b) : Mise en œuvre de changements prévus dans un protocole approuvé de gestion des modifications – La mise en œuvre du changement nécessite des informations complémentaires > Absence de conditions à remplir. > Quatre éléments supportifs à verser : 1. Référence au protocole approuvé de gestion des modifications. 2. Déclaration contenant les informations suivantes: le changement est en conformité avec le protocole approuvé de gestion des modifications et les résultats de l’étude répondent aux critères d’acceptation indiqués dans le protocole. Autre déclaration selon laquelle l’évaluation de la comparabilité n’est pas requise pour les médicaments biologiques/immunologiques. 3. Résultats des études menées en conformité avec le protocole approuvé de gestion des modifications. 4. Version modifiée de la/des sections concernées du dossier. |
| Variation Type IB/B.I.e.5c) : Mise en œuvre de changements prévus dans un protocole approuvé de gestion des modifications – Mise en œuvre d’un changement pour un médicament biologique/immunologique > Absence de conditions à remplir. > Cinq éléments supportifs à verser : 1. Référence au protocole approuvé de gestion des modifications. 2. Déclaration contenant les informations suivantes: le changement est en conformité avec le protocole approuvé de gestion des modifications et les résultats de l’étude répondent aux critères d’acceptation indiqués dans le protocole. Autre déclaration selon laquelle l’évaluation de la comparabilité n’est pas requise pour les médicaments biologiques/immunologiques. 3. Résultats des études menées en conformité avec le protocole approuvé de gestion des modifications. 4. Version modifiée de la/des sections concernées du dossier. 5. Copie des spécifications approuvées de la substance active. | Variation Type IB/B.II.g.5c) : Mise en œuvre de changements prévus dans un protocole approuvé de gestion des modifications – Mise en œuvre d’un changement pour un médicament biologique/immunologique > Absence de conditions à remplir. > Cinq éléments supportifs à verser : 1. Référence au protocole approuvé de gestion des modifications. 2. Déclaration contenant les informations suivantes: le changement est en conformité avec le protocole approuvé de gestion des modifications et les résultats de l’étude répondent aux critères d’acceptation indiqués dans le protocole. Autre déclaration selon laquelle l’évaluation de la comparabilité n’est pas requise pour les médicaments biologiques/immunologiques. 3. Résultats des études menées en conformité avec le protocole approuvé de gestion des modifications. 4. Version modifiée de la/des sections concernées du dossier. 5. Copie des spécifications approuvées du produit fini. |
L’utilisation du PACMP est fortement encouragée par les autorités réglementaires et en phase avec les dernières lignes directrices de l’ICH (Q8, Q9, Q10, Q12 et Q14). De ce fait, un accroissement de ce type de variations semble certain pour les années à venir.
Article rédigé par Isabelle MOUVAULT, Consultante Sénior en Affaires Pharmaceutiques