Mise en œuvre d’ICH Q12 en Europe : où en sommes-nous ?
En quoi consiste la ligne directrice ICH Q12 ?
La ligne directrice ICH Q12 « Guideline on technical and regulatory considerations for pharmaceutical product lifecycle management » vise principalement à fournir un cadre facilitant la gestion des modifications post‑approbation relatives au Module 3 du dossier d’Autorisation de mise sur le marché (AMM), ou modifications « CMC », de manière plus prévisible et plus efficace.
Cette ligne directrice et ses annexes ont été adoptées par le Comité des Médicaments à Usage Humain (Committee for Medicinal Products for Human Use – CHMP) de l’EMA en janvier 2020.
Ce texte tend à promouvoir l’innovation et l’amélioration continue, et à renforcer l’assurance qualité et la fiabilité de l’approvisionnement des produits, y compris la planification proactive des ajustements de la chaîne logistique dans un contexte de transparence entre l’industrie pharmaceutique et les autorités de santé.
Il s’efforce également de promouvoir, pour les régulateurs (évaluateurs et inspecteurs), une meilleure compréhension des systèmes de management de la qualité (SMQ) pharmaceutique des demandeurs pour la gestion des modifications CMC post-AMM en vue de minimiser les variations réglementaires dans un contexte international.
Enfin, il complète les lignes directrices ICH Q8 à ICH Q11 offrant des opportunités pour une approche basée sur la connaissance scientifique et le risque pour évaluer les changements tout au long du cycle de vie du médicament :
- ICH Q8 « Pharmaceutical Development » / ICH Q11 « Development and Manufacture of Drug Substances (Chemical Entities and Biotechnological/Biological Entities) » : se concentrent sur les premières phases du cycle de vie du produit (développement, enregistrement et lancement) ;
- ICH Q9 « Quality Risk Management » (QRM) : décrit les principes et les outils pour la gestion du risque qualité pouvant être appliqués à différents niveaux ;
- ICH Q10 « Pharmaceutical Quality System » (PQS) : décrit un modèle évolutif d’un système de gestion de la qualité pouvant être mis en œuvre tout au long du cycle de vie du produit.
ICH Q12 vise à démontrer comment une meilleure connaissance des produits et des procédés peut contribuer à déterminer plus précisément quelles modifications post‑approbation nécessitent effectivement une soumission réglementaire.
Pour ce faire, la version finale du texte adoptée par l’ICH en novembre 2019 présente 4 outils réglementaires et 4 facilitateurs réglementaires, accompagnés de principes directeurs, destinés à harmoniser au niveau mondial la gestion des modifications CMC post‑AMM (cf. Tableau 1).
Tableau 1 : Outils et des facilitateurs réglementaires ICH Q12
| Outils réglementaires | Catégorisation des modifications CMC postérieures à l’autorisation | Facilitateurs réglementaires | Système Qualité Pharmaceutique (PQS) et Gestion de des Changements |
| Conditions Etablies (ECs) | Lien entre l’Evaluation Réglementaire et l’Inspection | ||
| Protocole de Gestion des Modifications Après Autorisation (PACMP) | Approches Structurées pour les Modifications CMC Après Autorisation Fréquentes | ||
| Document de Gestion du Cycle de vie du Produit (PLCM) | Approches en matière de Données de Stabilité pour Soutenir l’Evaluation des Modifications CMC |
Vers une mise en œuvre facilitée des pleines possibilités offertes par ICH Q12
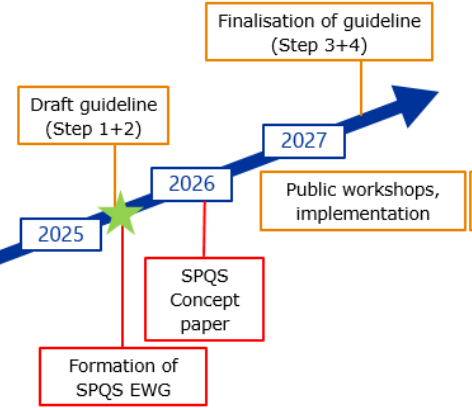
En mars 2020, soit deux mois après l’adoption de la ligne directrice ICH Q12 par le CHMP, la Commission Européenne et l’EMA ont publié une note explicative relative à la mise en œuvre avec des restrictions dans l’Union Européenne (i.e. l’ICH Q12 Step 5).
Au moment de la publication de cette note, des différences conceptuelles entre le contenu de l’ICH Q12 et le cadre juridique de l’UE ont été identifiées comme pouvant représenter un frein à la mise en œuvre totale de cette nouvelle ligne directrice en Europe ; ces différences ne permettant pas une application complète de la flexibilité opérationnelle et réglementaire définie dans l’ICH Q12.
En particulier, les approches scientifiques supplémentaires fondées sur le risque pour définir les conditions établies (Established Conditions – EC, cf. chapitre 3 de l’ICH Q12) et les catégories de notification associées, ainsi que le document de gestion du cycle de vie du produit (Product Lifecycle Management – PLCM, cf. chapitre 5 de l’ICH Q12), n’étaient pas considérées comme compatibles avec le cadre juridique l’UE relatif aux variations* en vigueur à cette époque.
Suite à la révision du règlement sur les modifications applicable depuis le 1er janvier 2025 en Europe, la Commission européenne a adopté et publié le 22 septembre 2025 la version finale des lignes directrices sur les détails des différentes catégories de modifications et le fonctionnement des procédures (applicables à partir du 15 janvier 2026). Ce règlement est ainsi désormais compatible avec l’ICH Q12 pour les sujets le concernant (cf. Tableau 2).
En ce qui concerne le système de qualité pharmaceutique (Pharmaceutical Quality System – PQS, cf. chapitre 6 de l’ICH Q12) et la gestion des changements, ainsi que le lien entre l’évaluation réglementaire et l’inspection (cf. chapitre 7 de l’ICH Q12), certaines clarifications supplémentaires concernant la démonstration et l’évaluation de l’efficacité du système de management de la qualité (SMQ) ainsi que la communication entre autorités réglementaires nécessitent encore une réflexion plus approfondie lors de la mise en œuvre de la ligne directrice (cf. Tableau 2).
Tableau 2 : Evolutions réglementaires européennes versus principaux outils/concepts ICH Q12
| Principaux outils/concepts – ICHQ12 | Ligne directrices variations (2013) | Ligne directrices variations (2025) | Commentaires |
| Chapitre 2 – Catégorisation des Modifications CMC Postérieures à l’Autorisation | Lignes directrices 2025 : ajout d’outils réglementaires additionnels compatibles ICH Q12 (QbD, PACMP, PLCM) dans cat. Q.I.e & Q.II.g | ||
| Chapitre 3 – Conditions Etablies (ECs) | Les ECs reflètent généralement les informations et caractéristiques de qualité soumises à variation. | ||
| Chapitre 4 – Protocole de Gestion des Modifications Après Autorisation (PACMP) | Lignes directrices 2025 : > Substance active : cat. Q.I.e.2 à Q.I.e.5 > Produit fini : cat. Q.II.g.2 à Q.II.g.5 | ||
| Chapitre 5 – Document de Gestion du Cycle de vie du Produit (PLCM) | Lignes directrices 2025 : > Substance active : cat. Q.I.e.6 à Q.I.e.8 > Produit fini : cat. Q.II.g.6 à Q.II.g.8 | ||
| Chapitre 6 – Système Qualité Pharmaceutique (PQS) et Gestion de des Changements | – | – | cf. ICH Q10 (PQS) & BPF (chapitre 1) – clarifications attendues |
| Chapitre 7 – Lien entre l’Evaluation Réglementaire et l’Inspection | – | – | clarifications attendues |
| Chapitre 8 – Approches Structurées pour les Modifications CMC Postérieures à l’Autorisation Fréquentes | – | – | clarifications attendues |
| Chapitre 9 – Approches en matière de Données de Stabilité pour Soutenir l’Evaluation des Modifications CMC | – | – | cf. draft ICH Q1 – clarifications attendues |
Compatible //
Pas compatible // – Pas de référence
Pour conclure, les évolutions de la réglementation européenne tendent à rendre effective la mise en œuvre de l’ICH Q12 afin de bénéficier de l’ensemble des progrès envisagés dans ce texte et ainsi faciliter la gestion des modifications CMC postérieures à l’autorisation de mise sur le marché de manière plus prévisible et plus efficace.
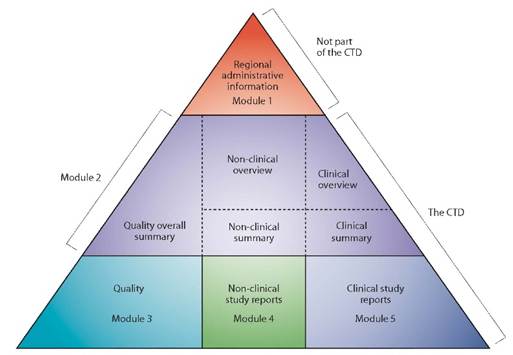
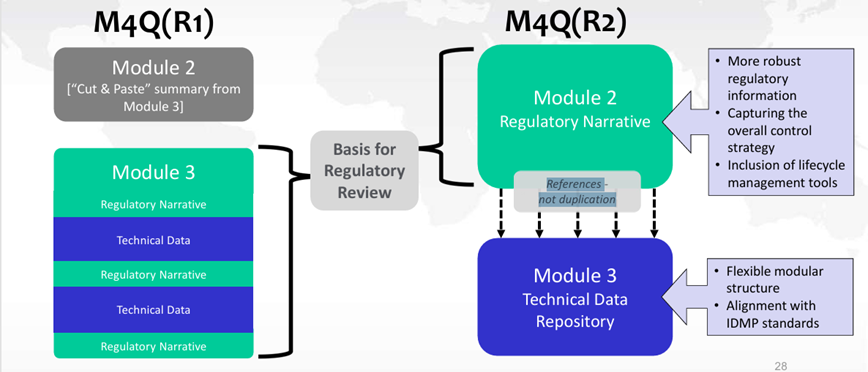
Par ailleurs, une future révision de la ligne directrice ICH Q12 sera à prévoir une fois la ligne directrice ICH M4Q(R2) « CTD on Quality » adoptée par le CHMP en Europe afin notamment d’en modifier l’annexe 1 (relative aux sections du CTD contenant des ECs).
* EC Variations Guidelines (2013) = Lignes directrices relatives aux caractéristiques des différentes catégories de modifications, au déroulement des procédures prévues aux chapitres II, II bis, III et IV du règlement (CE) n o 1234/2008 de la Commission du 24 novembre 2008 concernant l’examen des modifications des termes d’une autorisation de mise sur le marché de médicaments à usage humain et de médicaments vétérinaires et à la documentation à soumettre en vertu de ces procédures (2013/C 223/01)
** EC Variations Guidelines (2025)= Lignes directrices relatives aux caractéristiques des différentes catégories de modifications, au déroulement des procédures prévues aux chapitres II, II bis, III et IV du règlement (CE) no1234/2008 de la Commission concernant l’examen des modifications des termes d’une autorisation de mise sur le marché de médicaments à usage humain et à la documentation à soumettre en vertu de ces procédures (C/2025/5045)
Sources :
– ICH – Quality Guidelines
– EMA – Guidance on the application of the revised variations framework
Article rédigé par Isabelle MOUVAULT, Consultante Sénior en Affaires Réglementaires CMC